A recent article [Mer93] has examined the utility of introducing robot-like positional control into the field of synthetic chemistry to achieve molecular manufacturing, which will enable the construction and synthesis of an unprecedented array of desirable materials, including diamond and molecular computers. That article discusses what components and subsystems are needed to obtain a self-contained diamondoid manufacturing system, termed an "assembler", which is capable of synthesizing, among other things, copies of itself.
But as we do not yet have such a technology, naturally the question arises as to what pathways would lead from today's chemistry to the anticipated molecular manufacturing. This problem of putting together a self-contained system from a cruder level of sophistication by hooking up a number of more primitive parts is often encountered in computer science, and is termed "boot-strapping".
As diamondoid materials put many constraints on their synthesis procedure because they are so highly cross-linked and have such a high bond-density, they probably will have to be synthesized with highly reactive carbon species such as radicals and carbenes, and in a local high vacuum, which are conditions rather far from ordinary laboratory chemistry.
So in order to facilitate the boot-strapping process, the first primitive assembler which is able to self-replicate, should be constructed using less dense materials, more comparable to the polymeric species which are traditionally dealt with by liquid-phase chemistry. It has been shown in chapt.16 of [Dre92] that once a primitive assembler which achieves positional control to atomic precision has been constructed, migration pathways can be found that lead to diamondoid machinery through a few generations of increasingly sophisticated assemblers.
A primitive polymer-based assembler is still a large molecular aggregate by most of today's standards, with linear dimensions on the order of 100nm, somewhat larger than a ribosome. A description of what such an assembler might look like can be found in chapt.16 of [Dre92]. This stretches synthetic organic chemistry towards very large structures, while still having to maintain atomic precision. Many techniques like convergent synthesis and convergent self-assembly will have to applied in well thought out ways to be able to construct a properly functioning molecular aggregate this large. The following discusses a number of available options to meet this formidable yet intriguing and inspiring challenge.
To build such large molecular aggregates and components of machinery, one will want to rely to a certain degree on Brownian self-assembly of smaller sub-units, a principle that is observed throughout biochemistry. These sub-units should possess designed, complementary surfaces, so that self-assembly proceeds in a highly specific fashion by aligning the electrostatic interactions, hydrogen bonds, and the van der Waals contacts (combined with steric exclusion), which together make up the patterns of the complementary surfaces between pairs of sub-units. These sub-units have linear dimensions on the order of about 3nm, which is roughly the category of medium-sized proteins. The main problem therefore is to find a reliable and convenient procedure to fabricate a large diversity of such sub-units with clearly specifiable surface features.
The first class of compounds that springs to mind which would provide suitable sub-units, are the ordinary proteins themselves, and indeed this was proposed in the earliest description of the molecular manufacturing concept [Dre81]. Advances in protein engineering will eventually make it possible to create novel proteins based on the usual arsenal of amino acids, and which could become part of a primitive but general purpose assembler. This would essentially amount to being a flexibly programmable enzyme. Even though encouraging results in de novo protein design have been achieved [Ric92], progress has been slow, mainly due to difficulties in design (not in synthesis, which can often be easily accomplished by introducing the artificial DNA sequence for the protein into cellular expression systems with the help of a suitable vector, and then isolating the resulting protein).
The protein folding problem has not yet been solved: one still can not predict the tertiary 3D structure into which the long, one-dimensional chain of amino acids folds up, if one knows only the primary amino acid sequence and does not have the guidance provided by an already solved structure of a very closely related protein. This is so difficult because of the enormous number of bonds which allow torsion, leading to a combinatorial explosion of possible conformations, which can not be effectively dealt with even by large supercomputers applying a straight-forward brute force search. Much more subtle strategies would be necessary, the smart heuristics for which are difficult to find. Accordingly, most of what has been called protein engineering has restricted itself to introducing minor changes into proteins for which the 3D structures were already known, usually obtained by X-ray crystallography.
There exists an easier to solve, so-called reverse protein folding problem, which was first explained in [Dre81]. As the goal of constructing molecular machinery is rather an engineering than a scientific task, one has the freedom of exercising design choices. One can choose to only incorporate complexity to the degree humans can still clearly understand, which is a criterion which was not always respected by biological evolution. And so the task of finding an amino acid sequence which folds up in a predictable fashion should be much easier to accomplish than trying to find out how a sequence given by nature folds up, especially if one applies a systematic bias towards cutting down on complexity and conformational freedom. This area has not yet been explored very much and could be an important and fruitful path for further, relevant research.
Because ordinary proteins pose so many design difficulties, the temptation can hardly be resisted to gravitate towards the other extreme, to make the designing of the sub-units very simple, but to make their synthesis a more difficult problem. In this domain, one would like to build a 3D structure by joining molecular building-blocks which are able to form covalent bonds in three dimensions (as opposed to the essentially only one dimension offered by amino acids and nucleotides). One can imagine the picture of building a 3D crystal by stacking regularly shaped "bricks". This approach renders the design of a resulting sub-unit very easy, but shifts the problem to the task of finding the appropriate "bricks" and a satisfactory assembly technique.
In recent years there has been a rapidly rising interest in synthesizing large assemblies of organic molecules that might be able to serve as scaffolding structures in efforts to construct molecular objects of nanometer sized dimensions. An overview of the current direction of these investigations can be found in [Lehn93] and [Ama93]. Those molecular aggregates might find applications in molecular electronic and computing devices, or simply as novel materials with special chemical, optical, and electrical properties. These efforts have been an outgrowth of the field of supramolecular chemistry, which started with the serendipitous discovery that certain crown ethers are able to specifically complex (and so to "recognize") alkali metal cations [Ped67]. The resulting complexes were soluble in non-aqueous solvents, which seemed unusual for a compound that essentially is a salt. Analysis of the causes of this intriguing phenomenon has led to the synthesis of innumerable variations of these host-guest complexes [Cram88]. A very useful theoretical inquiry into the nature of self-assembly and molecular recognition of many similar complexation arrangements has been presented in [Lin91].
This growing research field is moving towards establishing an important enabling technology for the technological direction that has been outlined in [Dre81] [Dre86] and [Dre92], namely of eventually attaining manufacturing capabilities at the molecular level, leading to products which obtain their big utility from having all their atoms in precisely specifiable positions (as opposed to most of today's engineering materials like metals, ceramics, plastics, and wood, which have mesoscopically amorphous structures). It is useful to outline what molecular design issues in the field of supramolecular chemistry have to be systematically considered to help establish a focused effort to boot-strap molecular manufacturing.
Previous attempts directed towards finding a toolkit of elementary molecular units that can be stacked together in a flexible manner have been named largely according to brandnames of children's toys. Because the terms LEGO, Meccano, Tinkertoys, etc. already have their connotations of specific chemical implementations, I will refrain from using these occupied names and will simply call the abstract devices analyzed in this discussion "molecular building-blocks", or "MBBs" for short.
The largest amount of insight into finding relevant design criteria for MBBs is gained if one would like to employ them in a demanding application, like the construction of molecular machinery. Not only do the MBBs have to be able to stack in three dimensions potentially infinitely (to obtain scaffolding of large dimensions), but one also has to be able to specify distinct 3D stacking patterns and sequences, to obtain the highly idiosyncratic patterns present in machinery as well as in computers. As explained in [Mer93], achieving complex long range order is necessary.
For building machinery, it is necessary additionally to be able to specify the surfaces of interacting mechanical components in atomic detail, to construct required sliding interfaces (as discussed in chapt.10 of [Dre92]) and to place functional groups which can act as specific binding receptors or as catalytic sites similar to enzymes. In mechanical components, the ratio of interior volume to surface area is much smaller than in an infinitely extended regular crystal, because in these components mainly the surface gives a part its desirable characteristics. This being so, it follows that the slim interior of mechanical parts is best held together by strong interactions, preferably through the use of covalent bonds, to avoid the part from falling apart during usage. Most attempts in designing crystals and solids [Ama93] have so far only used ionic or even weaker interactions which would be unsuitable for achieving the declared goal of building molecular machinery. That is why covalent connections between the MBBs are assumed here.
Starting from a set of clearly stated questions [Kru91], subsequent analysis has found the following issues to be important in designing MBBs:
Hopefully the analysis of these two independent problems would result in two worked out "toolsets", which could then be combined flexibly in any desired fashion to give the actual incarnations of fully functionalized MBBs.
By exploiting symmetries in the MBBs and in the construction process, it is possible to mutually cancel irregular angles introduced by the particular chemistries of the links.
A very important question concerns the exact number of links that have to be established to integrate a MBB into a growing structure and in what sequence these links should be made. The first thing that comes to mind is that this number should be as low as possible. The lower the number, the fewer are the necessary chemical reactions and the higher is the yield of the final product (or maybe rather: the higher is the chance of getting a functional product at all, in the case of machinery). In addition, the difficulties of steric hindrance are somewhat relaxed if fewer links have to be established after the first one connects the newly added MBB with the structure. Maybe one could call this phenomenon a reduction of "link-density per volume" ?
In a very nice analysis of 3D-networks [Wel77] it is argued in chapt.4, that in order to form a regular 3D-lattice, a repeat-unit of this lattice has to have six free links for hooking up with neighbors, arranged in three pairs of diametrically opposed links, these link pairs being parallel to three non-coplanar axes, respectively. Unfortunately, it is very difficult to conceive of an organic molecule which would be able to undergo six link-forming reactions. Such a MBB would have to be an object with a cube-like geometry, and it is non-trivial to find an appropriate skeleton structure that would provide the required attachment geometries for the links.
But the argument in [Wel77] runs a step further: the repeat-units themselves can be built up from smaller parts, for example by joining two MBBs capable of forming four links each (leading to the diamond lattice in the case of tetrahedral geometry). Most currently investigated molecular aggregates, named "designer solids" in [Ama93], do not yet seem to have employed these lattice building concepts in a conscious design strategy. A good exception though can be found in [See82].
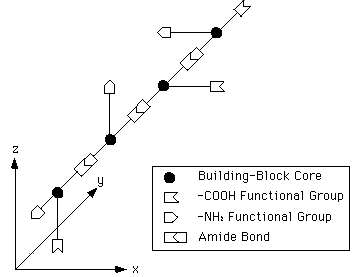
One could even use MBBs with only three links, and then four of them would be needed to construct a repeat-unit. This possibility is illustrated symbolically in figure 1.
FIGURE 1: A repeat-unit in a 3D lattice, composed of four smaller MBBs. The chemistry of the links indicated should just be taken symbolically.
A macromolecular structure would be assembled not by adding these rather large and elongated, inconveniently shaped repeat-units, but by adding the smaller constituent MBBs one at a time. On the average, one thus would have to form 1.5 links per block added, so one would alternate between forming one and two links per block. In this way, one has to go into the trouble of making two links at the same time only with half of all the MBBs used.
Here an important trade-off issue arises: the obviously beneficial desire to use MBBs with as few links as possible leads to more open and loose lattices which are less densely interknitted (lower link-density per volume) which also very favorably facilitates the access to individual bonds during construction. But because of the lesser crowding one will have to make sure that MBBs at the construction site, which are tied into the growing structure by so far only one completed link, do not obtain too many degrees of rotational freedom and twist away so that they still can be easily aligned for subsequent reactions. One way of solving this problem is to use link-chemistries which do not generate bonds that allow torsional freedom.
Also, the looser structures tend to show less mechanical rigidity, and the fairly large cavities in the interior of such lattices will be filled with solvent molecules instead of something that could contribute more to the stiffness of the structure. So it would be desirable to fill the empty spaces with some stuffing material which could increase the stiffness through non-covalent, van der Waals-type (vdW) interactions. This could be achieved by additional side chains that are attached to the MBBs and that pack in a definable way (see the further discussion on the CavityStuffer program below).
Because of the fundamental character of asymmetry, it is necessary to be able to also stack artificial MBBs in arbitrary ways that lead to chiral and asymmetric structures. Starting from MBBs which are chiral themselves, it is clear that this can be achieved, but it can also be accomplished by using achiral MBBs and introducing the asymmetry through the construction process by using a device that provides positional control (which in itself will always be a chiral entity!).
Macroscopic devices that provide positional control with atomic precision have been constructed now for many years, but they have been mainly used in an analytical mode to obtain high resolution images of all kinds of surfaces. An approach to using an atomic force microscope (AFM) in a constructional mode has been proposed in chapt.15.4 of [Dre92]. The strategy is to develop a rigid attachment procedure for antibody Fv-fragments, the smallest antibody fragments that still contain the full binding specificity, and on the AFM, both the flat surface which is traditionally the imaged part, as well as beads mounted on the cantilever tip are to be coated with these antibodies. This leads to a situation where there are antibodies on both sides of the AFM mechanism and can act as specific receptors. For this device, antibodies could be generated which bind small organic molecules having reactive functional groups, namely the MBBs, which will enable the mutual positioning of the reactive groups above each other to atomic precision, and to thus forge a chemical reaction at the desired location but nowhere else. The local effective concentration that can be achieved upon positioning the tip could be made to exceed 100M, and a background concentration of MBBs in the surrounding solution in the µM range is compatible with the affinities of antibodies. The difference in reaction rates between the selected reaction site and everywhere else should thus be of the order of 100,000,000. Such a large signal-to-noise ratio should allow the execution of at least 10'000 consecutive reaction steps on one substrate, which is unprecedented in organic chemistry.
Most types of reactions are not simply dimerizations but proceed in an asymmetrical fashion. This means that one usually can identify two differing chemical structures, one of which will connect onto the other. So one could say that the two components which will be joined to form a link are of two opposite abstract polarities. In an ideal case of link-chemistry, these two polarity components are each attached in the same way to the skeleton structure of a MBB, for example each using up one functionalization site. In this fashion, maximum design flexibility is obtained because it becomes possible to invert the polarity of a link between two MBBs in order to try out different assembly sequences and strategies without having to change anything in the MBB skeleton structures.
As link formation has to occur in such a crowded environment, the additional difficulty arises of planning for ejection trajectories for leaving groups, if the chosen reaction generates one. Also, for reactions with mechanisms which depend on interactions with the solvent, as is the case with reactions that need movement of protons, the detailed geometrical arrangement of solvent molecules in the vicinity of the reaction site would have to be known precisely. Such data is hard to get, and it probably can not be assured that the desired solvent arrangement will be present in the crowded space where links have to be formed.
A reaction which is in many ways quite ideal is the Diels-Alder cycloaddition, shown in figure 2.
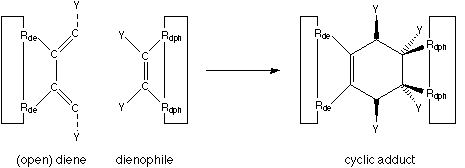
FIGURE 2: Diels-Alder cycloaddition.
The big benefits of a Diels-Alder reaction are: no leaving group is generated, nothing ionic is involved (which means that the reaction rate is little influenced by the dielectric constant of the surrounding environment), and it works in solvents from hexane to water. The two components of the Diels-Alder reaction can be viewed as two parts which simply snap together when held in sufficient proximity. The reactivity can be tailored by the appropriate attachment of electron-withdrawing and electron-supplying neighbouring functional groups (denoted as "Y" in figure 2).
But a big disadvantage is that the two reactive polarities involved, a diene and a dienophile, can not be dealt with like ordinary (heteroatomic) functional groups. Depending on the design, they are likely to end up not being symmetrically exchangeable because the two polarities most often have to be anchored differently on a MBB skeleton. Incorporating these two functionalities creates a lot of trouble as usually at least two functional group attachment sites would have to be used up, and maybe the whole skeleton structure would have to be custom-tailored around these reactands. This would violate modularity boundaries and sacrifice the flexibility gained by being able to separately design skeleton structures and the functional group decoration.
On the other hand, these functionalities offer quite some flexibility in decorating the formed cyclic adduct by additional functional groups, denoted by "Y" in figure 2, that may serve for more than just the fine-tuning of reactivity, and which could turn the links between MBBs into structure-contributing entities in their own right.
But by ordinary synthetic chemistry standards this represents a pathologically low concentration of material and the synthesis and storage of MBBs is almost certainly going to occur at quite higher concentrations, very likely in the mM range. But in turn this then means, if the MBBs have the same potential reactivity all the time, that the rate of random reactions jumps by a similar factor, namely about thousandfold. And so the raw materials would get degraded at an uncomfortably high rate by mere storage, before they ever even get to the scene of action. Clearly, this problem has to be addressed. Several potential solutions are thinkable.
Besides techniques like cryogenic storage and storage in an inactive or protected form, the presumably simplest method is the storage of only compatible classes of chemical polarities.
As the stability problem in storage arises mainly because MBBs will in the usual case have link-formation functionalities of both polarities (as e.g. amino acids do too), which enables them to react with each other and polymerize spontaneously, the problem could be solved by somewhat restricting the freedom of functionalization patterns. MBBs could be separated into different distinct classes of linking-chemistries, which contain functional groups of only one type of polarity that is not able to dimerize. This segregation of MBBs into classes of mutually compatible chemical polarities will limit the flexibility of inverting the polarity of links and produces more complicated constraints for designing and assembling macromolecules. But the big benefit will be on the practical side with much easier handling of the raw materials. Essentially no special precautions would have to be followed.
It might be asked why one should worry at all about the skeletons of MBBs as it should be no big deal to chunk together a few appropriately functionalized rigid molecules. And even though these ad hoc chosen molecules might have rather irregular geometries, it still would be possible to obtain clean and regular rasters in the structure-to-be-built by adhering to the exploitation of symmetries in the construction process which can cure out and cancel irregularities in the individual MBBs. But just a casual look at the available repertoire of molecules and a more detailed look at potential MBB-candidates renders a more bleak image.
As MBBs are to be connected in a three-dimensional fashion, one needs to find rigid and three-dimensional molecules which could serve as skeletons. But apart from the fact that the overwhelming bulk of molecules that chemists deal with are of a floppy and chain-like nature, the few rigid and compact cages (like cubane, adamantane, dodecahedrane, the norbornanes, and the fullerenes) are very difficult to functionalize in a systematic and useful way because these molecules are very inert once synthesized. The step-wise syntheses of these skeletons often face severe steric problems at one or more steps, they often have lengthy syntheses, give low yields, and harsh conditions are employed which many functional groups would not tolerate. The most promising structures would be the norbornanes, which usually are assembled in one step by a Diels-Alder cycloaddition reaction, starting with two substrate materials that could be functionalized with some limited degree of flexibility.
But even if this group of compact cage skeletons were easily accessible, one could raise doubts on their usefulness because they might be too small for practical purposes. They might not provide enough potential attachment sites for functional groups. One of the more interesting types of chemistries to link the MBBs together are electrocyclic additions of the Diels-Alder type. The problem there would be that in order to just anchor one link, one would have to occupy two adjacent functional group attachment sites, at least for the diene component. With small skeletons such as adamantane and norbornane, one would encounter severe problems in incorporating diene structures.
The small sizes of the compact cages could also make them not immunogenic enough, which is of importance because the receptors for these MBBs will presumably be antibody fragments. On the other hand, antibodies to a hapten of comparable smallness, 2,4-dinitrophenolate, have been successfully generated and are commercially available.
If the compact cage skeletons are too small, one would simply have to use larger skeleton structures. But what are the problems one runs into here ? In the literature, there is an intriguing lack of syntheses of stiff larger cage structures that might be suitable. There is quite a range of cavitands, carcerands, and similar complexing agents, but the functional groups of these compounds generally all look towards the interior, and/or are chemically identical and indistinguishable, and are provided in insufficient quantity (many large compounds hardly have three or four functionalities).
Ideally, one would like to have a dedicated construction kit for the MBBs themselves, to build a variety of them, having different and asymmetric functionalization patterns. This kit would consist of a number of miniature building-blocks, each able to carry, say, one decorational functional group that is to end up in the final MBB, plus functional groups needed to link up with other minis to establish the skeleton structure. Such an approach has been tried recently with some success [WuLeeMoo92] by using phenylacetylene-units as the minis, which can be joined in a sequentially controllable polymerization scheme. In principle, one could custom-tailor the phenyl-units to contribute decorational functional groups.
In order to create stiff, non-floppy, cage-like structures, one invariably will end up pinning down a potentially flexible "planar" structure by a tripod. This whole construct will end up being rigid and sturdy only if the legs of the tripod do not contain too many joints. Namely, one joint seems to be fine as is nicely illustrated by adamantane, where a "planar" cyclohexane ring is being held in one conformation by a tripod which has one joint per each leg. (Cubane is a "planar" cyclohexane ring being held in one conformation by two tripods which have zero joints in the legs and are grasping the cyclohexane from both sides and holding at it alternating sites.) But with more than just one joint per leg, the tripod will become floppy. Each joint will be where an atom sits, and this statement can be also reversed: as there are no atomic bonding geometries in organic chemistry that are linear (except for sp1-carbons), every atom will introduce angles and become a joint that is able to wiggle the leg into undesired conformations, unless strictly confined, by e. g. a tripod that does not have legs which contain more than one intervening joint. Building sturdy structures using these minimal tripods invariably leads to cyclic structures that contain about six atoms, which is precisely the domain of the compact cages mentioned earlier. So then, nothing would have been gained. It is fairly difficult to conceive of larger cages that can be put together in a modular fashion so that the result is still rigid enough. In [WuLeeMoo92] the large cage was constructed by not using single atoms as the joints, but phenyl-units instead. The architecture used might be described as two mutually opposed tripods, which seem to be able to restrict each others conformational freedom sufficiently in some cases. This however led to a fairly empty and airy cage which leaves some desire for more structural rigidity.
There is an additional problem. As any individual mini-building-block has to be incorporated into the skeleton in a rigid and confined fashion, it usually will have to be attached by at least three bonds. If this mini then provides one additional functional group that will actually appear on the MBB, then in the overall analysis, three functional groups have been consumed for one that has been delivered. If one looks at the totally and completely assembled MBB, one finds that essentially nothing has been gained in terms of providing more potential functionalization sites which would have been desirable for increased design flexibility, and which was the reason why one wanted larger cages in the first place. The functional groups needed for tying together the minis into the skeleton are lost because usually the atoms for such a bond cannot engage in any further activity other than the bond-formation itself. Similarly, nothing is gained in additional functionality by using sp1-carbons (the only linear joint type available as mentioned above), because the resulting acetylenic structures only make the cage larger and more airy.
These problems seem to constitute a rather sinister law in organic chemistry which explains in part why modularily enhancing the number of functionalization sites by making larger (but still rigid) cages fails so easily. After more than a hundred years of synthetic efforts, one would have expected large cages to be more prevalent unless there do exist serious obstacles.
An interesting and helpful structural unit in the design of such "integrated" MBBs can be found in using spiro-connections, i. e. two rings sharing exactly one carbon-atom. By using these, one is able to obtain a rigid 90 degree orientation of two planar structures in the smallest possible volume.
![]()
FIGURE 3: Chemical bonds acting as spacers are generating large cavities in a lattice.
This empty space does not contribute to the structural stiffness of the material, being filled with solvent molecules, which of course are too fluid to achieve significant mechanical strength. It would be very desirable to bring the cores of the MBBs together much closer, so that their mutual vdW-interactions are able to contribute to the material stiffness. This is the case in the interior of proteins, where most of the stability comes only from weaker-than-covalent interactions. But then on the other hand, the internal design of proteins does not adhere very much to well organized rasters, which renders their design much more difficult. A strategy that might be pursued to reduce the spacer-effect is illustrated symbolically in figure 4.
![]()
FIGURE 4: Reducing the adverse separation effect of the spacers.
The idea is to fold back the link-chemistry onto the surface of the MBBs, so that the links do not stand off like the spines of a hedgehog. An actual implementation might look like figure 5.
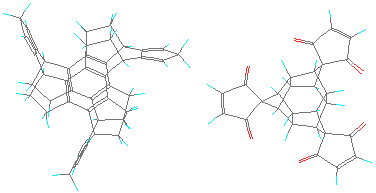
FIGURE 5: A pair of MBBs with three-fold connectivity. The two components trindan-diene and iceane-tridiph can be retrieved for viewing in a molecular modelling program.
Note that this proposed design also incorporates the separation of the link-polarities to circumvent the storage problem alluded to above. That is why there are two distinct types of MBBs here. Only the one on the left side bends back the link-functionalities somewhat. This example demonstrates the new problem with this approach. To bend back the links, it is necessary to introduce more angles in the skeleton, which have to be rigid to avoid floppy structures. This leads to polycyclic structures which are increasingly difficult to synthesize (without already having access to molecular manufacturing methods :-).
Surprisingly, a tendency in current research seems to have turned the cavity problem into a virtue. Design of zeolites has become very popular, because of the hope that the large internal surface areas might be used for catalytic or separational purposes. But the zeolite designers are encountering a recurring problem which is again related to the emptiness of the very cavities they would like to create. Very often not one lattice is generated, but several, which are independent but mutually interlocked, and so all of the space is filled out nevertheless. An instructive example can be found in [Erm88]. This can be avoided by the following trick. During the growth of the zeolite crystals, one has to provide template molecules which are able to claim the cavity-space, to prevent the formation of the interpenetrating lattices. One of the most successful attempts and a concise description of the zeolite phenomenon can be found in [HosRob90]. But the issues involved in getting useful zeolite designs to work, add an additional twist to the already significant challenge of designing good MBBs.
It seems as if the real challenge in constructing designer solids lies in achieving dense packing. Airy zeolite-like structures have been prepared several times, accidentally and deliberately. But the systematic elimination of the empty spaces is the difficult task. This situation is reminiscent of the challenges in the protein engineering community, where the first step of getting artificial proteins to fold up into roughly the right way, including secondary structures, seems to have been easier than commonly imagined, but the last step of getting the amino acid side chains to organize properly in the interior to obtain really good tight packing, seems to be unexpectedly hard to accomplish [Ric92].
A number of rod-like molecules have been synthesized [KasFriMic92] [YanEtal92], the ends of which could be connected in suitable hubs. But this would lead to the zeolite problem in its most extreme form. The longer the rods, the more humungously large the cavities become. Also, most rod proposals do not have handles, or flexible functionalization possibilities, and so the problem of assembling useful and arbitrary structures with them is still unsolved. Building extended structures and lattices using these building blocks has yet to be demonstrated.
Successful attempts at deliberate lattice building have been accomplished [SimSuWue91] by using tetrahedral MBBs that assemble into a diamond-like lattice. But the crystal is held together only by weak hydrogen bonds, and no actual skeleton-design was pursued. Also, the functionalities forming the links have a rotational degree of freedom, which renders the exact arrangement in the crystal unpredictable.
An interesting approach to MBB skeleton-design has been followed in [WuLeeMoo92], leading to a skeleton construction toolkit with phenylacetylene-units, as mentioned above. This scheme introduces a number of potential functionalization sites, but those have not yet been exploited for achieving specific intermolecular aggregation and thus predictable lattice-formation.
Much work follows more closely the traditional kinds of host-guest chemistry and the self-assembly of compact and "introverted" entities, as surveyed in [Lin91]. For example, [KohMatSto89] describes a number of closed, self-contained structures which are not designed to assemble into larger lattices in a definable way. They do not really live up to the term Molecular LEGO which was used to describe some of the structures, because the proposed molecules lack the extendibility beyond a closed entity of finite size, which would be precisely the feature that gives LEGO its wonderful properties.
A complication arises through the need for positional control while assembling lattices with idiosyncratic stacking patterns. A modified AFM suitable for the task seems within short term experimental reach, but has not yet been demonstrated. Avoiding the necessity of introducing positional control can only be accomplished if the MBBs carry a sufficient amount of encoded distinguishing-information with them, implemented as surface patterns of functional groups. This could allow preprogrammed, structure-directed self-assembly to take place.
Obviously, the amount of information which can be encoded depends on the surface area available, which strongly favors large MBBs. Also through large surface areas, the strength of intermolecular interactions with neighbouring MBBs can be made suitably high, if one does not want to rely on covalent links. (Arguably the largest link-strength per cross-sectional area can be obtained by covalent bonds.) To obtain a feel for how large the area of contact between a pair of MBBs should be for good strength and specificity, one can imagine unfolding and flattening out the convoluted surface of an enzymatic binding site that recognizes a small molecule. The unfolded surface area is uncomfortably large. Trying to devise MBBs in this size range causes a lot of problems in terms of structural rigidity and very complex syntheses.
At both extremes of the conceptual design space we see difficult obstacles. On one hand there are natural proteins which are easily synthesized but are very difficult to design, on the other hand there are lattices of MBBs which are easy to design but problematic to synthesize and assemble. What has not yet been sufficiently explored is the territory in between these extremes. A trade-off can be made along the lines of the reverse protein folding problem. Synthesizing a largely linear or slightly cross-linked polymer is not too difficult. But one would like to increase the designability by using unorthodox amino-acid-like moieties which, through the bulkiness and rigidity of their artificial side chains, are able to restrict the conformational freedom of the folding polymer considerably. This can substantially reduce the combinatorial explosion of different conformations and thus the search space that has to be considered to find satisfying solutions. In this domain, design is not as intractable as in natural proteins, but is nevertheless more difficult than humans would want to be confronted with directly. To assist the human designer, computer-aided design tools are necessary that are able to autonomously generate proposals for polymers that fold into a tightly packed structure.
The author is currently working on such a program, called CavityStuffer. It generates its proposals by "growing" polymers blindly within a given volume, the surface of which has been prescribed to atomic precision. In a randomized fashion, it chooses sites on the tree-like polymer for further attachment and elongation. Once a site has been chosen, a moiety (monomer) is selected from a library of allowed moieties, similar in concept to allowed moves in a game of chess. The moieties are stored in the library as rigid, three-dimensional puzzle-pieces (as rotamers); so the CavityStuffer program works by trying to fit three-dimensional geometric shapes into the given volume. Whenever a chosen moiety does not fit (i.e. if it clashes with other atoms already in place), it is discarded, and a different moiety will be tried instead. This scheme largely avoids the expensive numerical tasks encountered in energy calculations (which are commonly used for studies of protein folding), because clash-detection is quick. Because the generation of individual proposals is fast, a large number can be generated automatically without human super-vision and sorted according to how well they fill the volume. There are various places in the program where one could add in smarter and higher-level strategies than the random selection mechanism used now.
Currently the basic machinery of the CavityStuffer program has been implemented and is working. One can let it build tree-like molecules using moieties provided by a library. There are also tools to help construct the moiety-library, and there are facilities for input and output via PDB files. But with a simplistic demo-version of the moiety-library, the packing of the polymer is not yet very good. More work has to go into a careful design of good moiety-libraries which contain a high enough diversity of rotamers. Better packing strategies need to be added as well. Interested readers who would like to stay informed on this project are welcome to send e-mail to kr@shell.portal.com.
Much of what has been discussed here has a strong flavor of engineering, but at the same time provides directions where new and fruitful scientific playgrounds may be found.
By analyzing the new aspect of chemistry which is dealing with specific intermolecular aggregation, one will learn the design rules by which smaller building-blocks can be assembled into supramolecular entities to achieve a "chemistry beyond the molecule" [Lehn93]. Rewards of this development will be many novel materials with special optical, electrical, and perhaps magnetic properties, which will have uses in their own right. These novel materials most often have fairly simple and regular lattice structures.
But the big payoff will come from using the insight gained from the intermolecular assembly strategies to build parts of molecular machinery, which are entities of distinct sizes and shapes, not just infinitely extended crystals. The goal is to use these machine parts to build a first crude assembler which is able to self-replicate, and to thus boot-strap molecular manufacturing.
Small organic molecules serving as molecular building-blocks have the problem that they are too small to encode significant amounts of information to allow their preprogrammed self-assembly into aggregates with highly idiosyncratic structure. There is a lack of sound and reliable engineering guidelines for designing preprogrammed and autochthonous assembly reactions [Lin91], which is not surprising considering that small molecules provide so little encoded information available for manipulation. Therefore a positional synthesis device is needed which can assemble the materials under external program control.
A promising alternative for obtaining protein sized sub-units is the folding polymer approach, where some amount of the design complexity is mitigated through the use of rigid, non-standard amino-acid-like moieties. A large proportion of the remaining design task is off-loaded to automated design programs which are based on the packing of rotamers, similar in concept as described in [PonRic87]. It is indeed surprising that this existing rotamer approach has so far been used only for understanding natural proteins, instead of extending it for designing new ones. The full arsenal of available methods has not yet been unleashed on the problem of de novo protein engineering and the design of folding polymers. There is reason to believe that a more vigorous effort in this direction will be very successful.
This article is dedicated to Viviane Bracher. I thank the Institute for Molecular Manufacturing for financial support, and K. E. Drexler for many stimulating discussions. (Copyright 1993 by IMM. All rights reserved.)

send e-mail to:kr@shell.portal.com
(Copyright 1993 by IMM. All rights reserved.)
v. 1.4 of July 1996